
Finding Our Way with L1CAM
 The causes of hydrocephalus are varied and in many cases result from a combination of genetic susceptibility, environmental factors, and injury. One example with a clear genetic linkage is X-linked hydrocephalus, a rare genetic disorder that occurs in about 1 of 30,000 births (Edwards 1961). X-linked hydrocephalus is characterized by stenosis, or narrowing, of the aqueduct of Sylvius and severe ventriculomegaly. It is part of a group of conditions linked by mutations in the L1CAM gene. Collectively the conditions are called L1 syndrome, whose symptoms can vary greatly in severity, and include: MASA (mental retardation, adducted thumbs, shuffling gait and aphasia) syndrome, certain forms of X-linked spastic paraplegia (SPG1), and X-linked agenesis of the corpus callosum (ACC).
The causes of hydrocephalus are varied and in many cases result from a combination of genetic susceptibility, environmental factors, and injury. One example with a clear genetic linkage is X-linked hydrocephalus, a rare genetic disorder that occurs in about 1 of 30,000 births (Edwards 1961). X-linked hydrocephalus is characterized by stenosis, or narrowing, of the aqueduct of Sylvius and severe ventriculomegaly. It is part of a group of conditions linked by mutations in the L1CAM gene. Collectively the conditions are called L1 syndrome, whose symptoms can vary greatly in severity, and include: MASA (mental retardation, adducted thumbs, shuffling gait and aphasia) syndrome, certain forms of X-linked spastic paraplegia (SPG1), and X-linked agenesis of the corpus callosum (ACC).
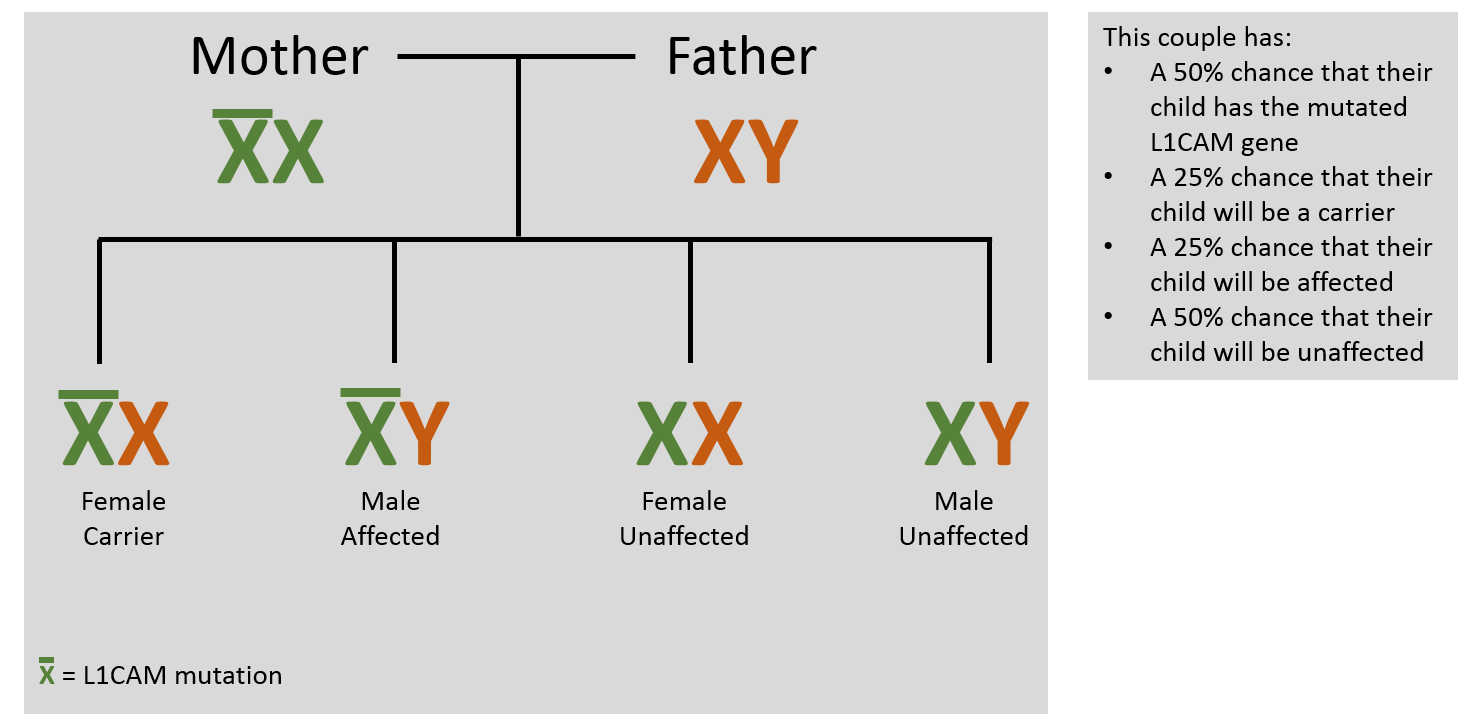
The vast majority (99%) of symptomatic individuals are male. L1CAM mutations can occur in a person through a spontaneous, new (de novo) mutation or by inheriting the mutation from his or her mother. There are no reported cases of inheritance through the father. L1CAM mutations are inherited through the maternal line because L1CAM is located on the X chromosome (X-linked inheritance). During reproduction, an individual acquires two sex chromosomes, one from each parent. Females have two X chromosomes (XX) while males have an X chromosome from the mother and a Y chromosome from the father (XY).
Females with one affected X chromosome are called ‘carriers’ and have less than a 5% chance of showing clinical symptoms. This is because females have a second, intact copy, of the L1CAM gene which is inherited from the father. A female carrier has a 50% chance of transmitting the affected gene to each child. Males with an affected X chromosome have L1 syndrome because the Y chromosome does not contain the L1CAM gene. Therefore, in males, all of the gene products are produced from the one affected L1CAM gene. The severity and type of symptoms are dependent on where and what genetic mutation is present.
During brain development, the L1CAM gene produces the L1 cell adhesion molecule (L1). L1 is primarily present in developing neurons and plays a critical role in guiding new neurons into the correct positions and helping axons grow and make connections with other neurons. Disruptions caused by mutations in the L1CAM gene impair these critical function and alter brain development.
Recent studies have further elucidated the role of the L1CAM gene in neurodevelopmental processes. In addition to guiding neuronal migration and axonal growth, L1CAM is involved in synapse formation, dendritic spine development, and the maintenance of neural circuits (Tai et al., 2019). These functions are crucial not only during brain development but also for neural plasticity in adulthood. Mutations in L1CAM and resulting disruptions to these processes can cause neurons in the developing brain to be miss-positioned and fail to make the necessary connections to other neurons throughout the brain and spinal cord. These factors could account for clinical symptoms such as adducted thumbs, developmental challenges, and motor difficulties. Additionally, these disrupted connections can contribute to cognitive challenges and other neurodevelopmental issues observed in L1 syndrome.
An additional question is why disruptions in L1CAM cause hydrocephalus. The relationship between L1CAM mutations and hydrocephalus is still under investigation, but new insights have emerged. One theory suggests that the mutations may impair the function of ependymal cells, which line the ventricular system and play a role in cerebrospinal fluid (CSF) circulation. When these cells are not functioning properly, CSF flow can be disrupted, potentially leading to aqueductal stenosis and hydrocephalus. Another theory proposes that L1CAM mutations affect the development and organization of white matter, reducing brain elasticity and making it more susceptible to changes in intracranial pressure. These factors combined may increase the risk of hydrocephalus in individuals with L1 syndrome (Itoh & Fushiki, 2015).
Continued research is essential to better understand the mechanisms by which L1 dysfunction leads to these neurological abnormalities. By uncovering the specific roles of L1 in neural development, scientists hope to create targeted therapies that can prevent or halt disease progression. Future treatments may involve approaches such as gene therapy to repair or replace the defective L1CAM gene or L1 protein, potentially addressing the root cause before any harm can occur.
Here are some Research 101 blogs by Dr. Jenna Koschnitzy, our previous Research Programs Director, that provide good background reading to better understand this blog: